indhold:
- Medicinsk video: My Horibal Speling
- Hvordan kan siglcelle sygdom opstå?
- Kan siglcelleanæmi blive helbredt?
Medicinsk video: My Horibal Speling
Udtrykket seglcelle sygdom eller seglcelle sygdom (SCD) indbefatter flere arvede abnormiteter i røde blodlegemer. SCD-lider har unormalt hæmoglobin kaldet hæmoglobin S eller sigtehæmoglobin i deres røde blodlegemer.
Hæmoglobin er et protein i røde blodlegemer, der bærer ilt i hele kroppen.
Udtrykket "arv" indikerer, at denne sygdom er forårsaget af gener, der overføres af forældre til deres børn. I modsætning til forkølelse eller infektion er SCD ikke smitsom, så sunde mennesker ikke vil blive udsat for SDC fra lider.
SCD lider arve to unormale hæmoglobingener fra far og mor. Uanset hvilken type SCD der lider, får mindst en af de to abnormale gener kroppen til at producere hæmoglobin S. I mellemtiden vil en person, der har to hæmoglobin S-gener (eller SS-hæmoglobin), lide af en sygdom kaldet sickle-cellemæmi. Sickle celle anæmi er den mest alvorlige og mest almindelige type SCD.
SC hæmoglobin og hæmoglobin Sβ-thalassæmi er to andre almindelige former for SCD.
Nogle former for seglcelle sygdom:
- Hemoglobin SS
- Hæmoglobin SC
- Hæmoglobin Sβ0 thalassæmi
- Hæmoglobin Sβ+ thalassæmi
- Hemoglobin SD
- Hæmoglobin SE
Hvordan kan siglcelle sygdom opstå?
Kropsvæv i kroppen har brug for en iltforsyning til at fungere korrekt. Normalt tager hæmoglobin i røde blodlegemer ilt fra lungerne og bærer det til alle kropsvæv.
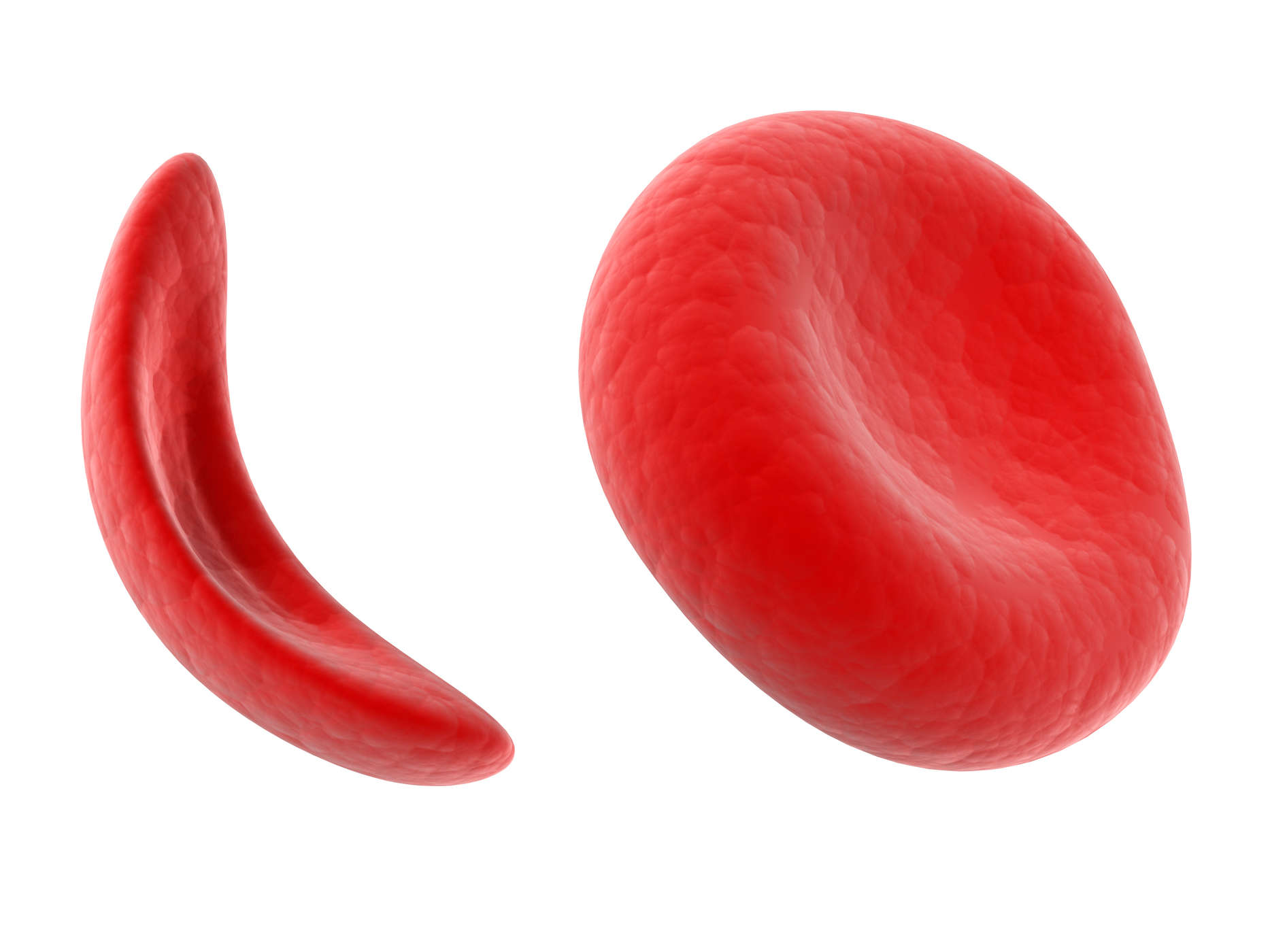
Røde blodlegemer, der indeholder normalt hæmoglobin, er formet som stykker (som donuts uden huller). Denne form gør det muligt for celler at blive fleksible, så de kan bevæge sig gennem store og små blodkar for at transportere ilt.
Sickle hæmoglobin er ikke som normalt hæmoglobin. Sickle hæmoglobin danner en stiv stamme i røde blodlegemer, bliver til en segl eller form segl.
Halvmåneceller er ufleksible og kan klæbe til kargen væggen, hvilket forårsager blokeringer, der sænker eller stopper blodgennemstrømningen. Når denne tilstand opstår, kan ilt ikke nå det omgivende væv.
Manglende vævs oxygen kan forårsage alvorlig smerte kaldet smerte krise (smerte kriser). Denne smerte kan pludselig ramme uden varsel, så patienterne behøver effektiv behandling på hospitalet.
De fleste børn med SCD oplever ingen smerte eller smerte krisermens unge og voksne kan lide af kronisk, igangværende smerte.
Unormale røde blodlegemer og lavt iltindtag kan også forårsage skade på organer, såsom milt, hjerne, øjne, lunger, lever, hjerte, nyrer, penis, led, hud eller endda knogler.
Fordi formen ikke er fleksibel, kan seglceller eksplodere eller kaldes hæmolyseres, Den normale livscyklus for røde blodlegemer varierer fra 90 til 120 dage, mens seglceller kun kan vare i 10 til 20 dage.
Kroppen gør altid nye røde blodlegemer til at erstatte gamle celler. Imidlertid har kroppen af SCD-lider problemer med at afbalancere ny blodcelleproduktion med den hurtige ødelæggelse af blodceller. Som følge heraf har antallet af røde blodlegemer tendens til at være lavere end det normale antal. Denne tilstand kaldes anæmi som en årsag til kroppens reducerede energi.
Kan siglcelleanæmi blive helbredt?
Sickle celle sygdom er en livslang sygdom. Alvorligheden af sygdommen hos hver lidelse skal variere.
I højindkomstlande som USA er levetiden for SCD-syge i øjeblikket omkring 40-60 år. I 1973 var gennemsnitsalderen for SCD-patienter i USA kun 14 år. Dette viser fremskridt i diagnosen og behandlingen af SCD.
På dette tidspunkt, hæmatopoietisk stamceltransplantation (HSCT) er den eneste medicin til SCD. Desværre er de fleste SCD-lider for gamle til at gennemgå en transplantation eller har ikke familiemedlemmer, der har en genetisk kamp som donor. Egnede donorer er nødvendige for at få de bedste transplantationsresultater.
Der er mange effektive behandlinger, der kan reducere symptomer og forlænge SDC-patientens liv. Tidlig diagnose og regelmæssig lægehjælp for at forhindre komplikationer bidrager også til at forbedre livskvaliteten for patienterne.